Advancing Genetic Engineering: Meet the first FDA-Approved cell-based Gene Therapies for treating Sickle Cell and its Implications for future Disease Treatment
Ezekiel Bajomo • 2024-06-30
𝙄𝙣𝙨𝙩𝙞𝙩𝙪𝙩𝙚 - 𝘽𝙞𝙤𝙡𝙤𝙜𝙮 𝙃𝙖𝙧𝙫𝙖𝙧𝙙 𝙐𝙣𝙞𝙫𝙚𝙧𝙨𝙞𝙩𝙮 Casgevy and Lyfgenia, developed by Vertex Pharmaceuticals and Bluebird Bio respectively, are the first treatments of their kind targeted at treating sickle cell anemia with one use. These breakthrough procedures hold much promise for the future of treating similar genetic diseases but also raise questions about costs, accessibility, and ethics.
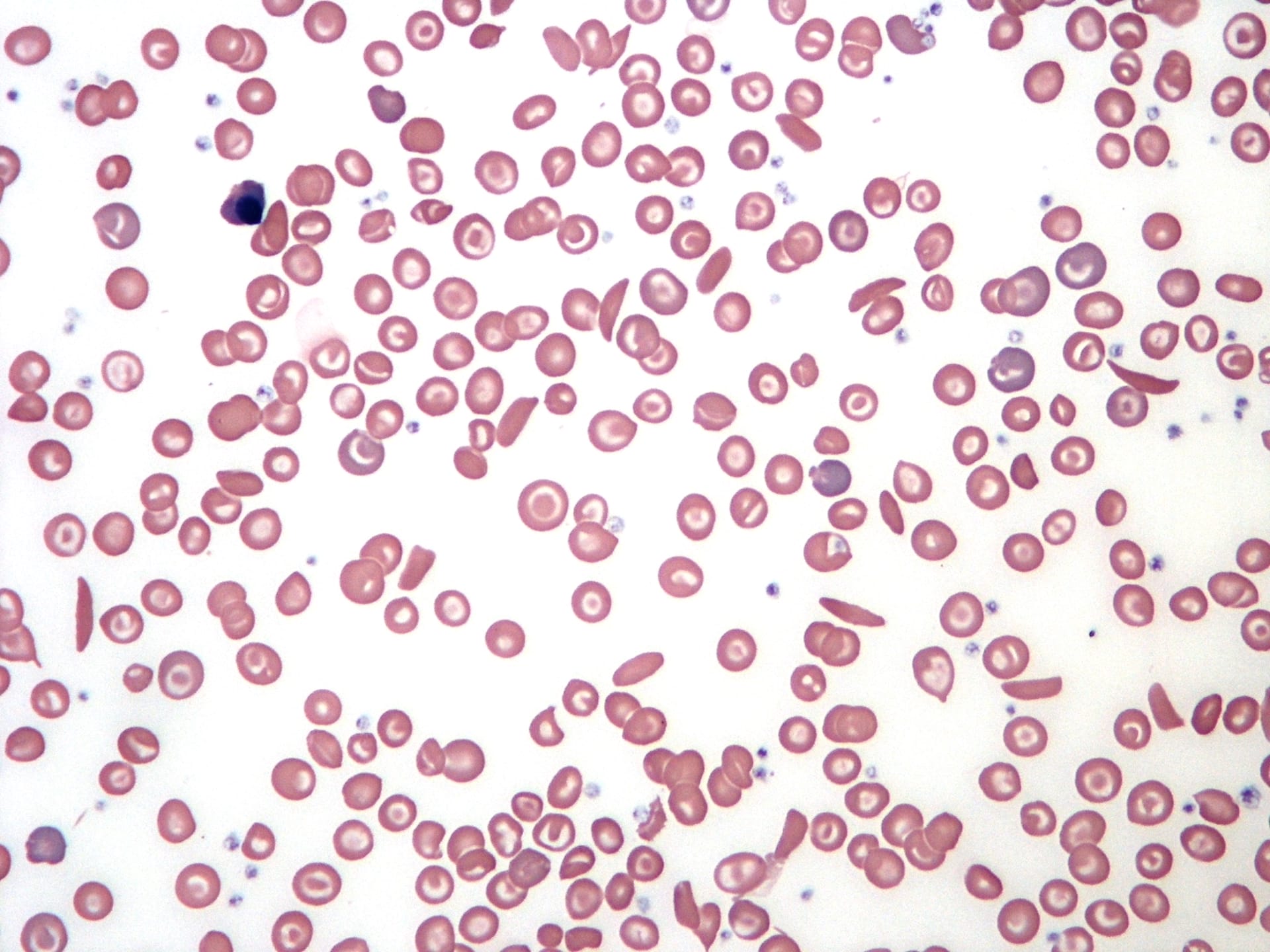
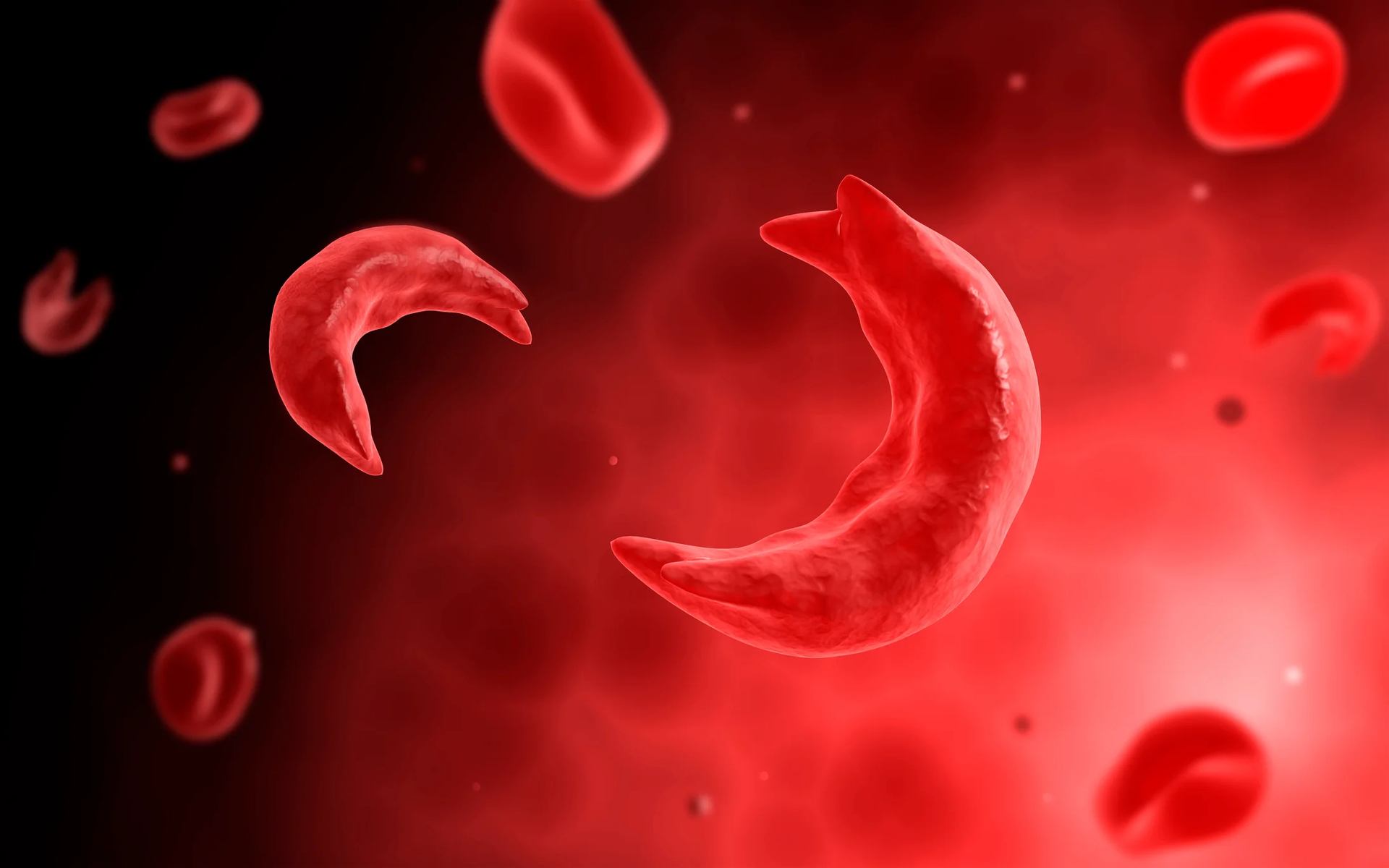
Figure I. Image of abnormally shaped red blood cells, also known as sickle cells, in the bloodstream.
Origin
The origin of sickle cell starts 7,300 years ago with the birth of a child in West Africa, a rainy and densely forested region with the perfect conditions for mosquitoes that carry malaria – a deadly disease that was estimated to have killed 150 to 300 million lives just in the 20th century according to the National Institute of Health. The child was born with a genetic disease that gave them an enhanced immunity against malaria. With this newfound imperviousness to the disease, the child went on to live a long healthy life and passed down the favorable genetic mutation over two hundred generations to the present day. However, there was a fatal problem with this genetic mutation: if two parents carry the trait, their child can inherit two copies of it, leading to Sickle Cell Disease (SCD). This disease causes severe pain, weakness, tiredness, and a multitude of other complications.
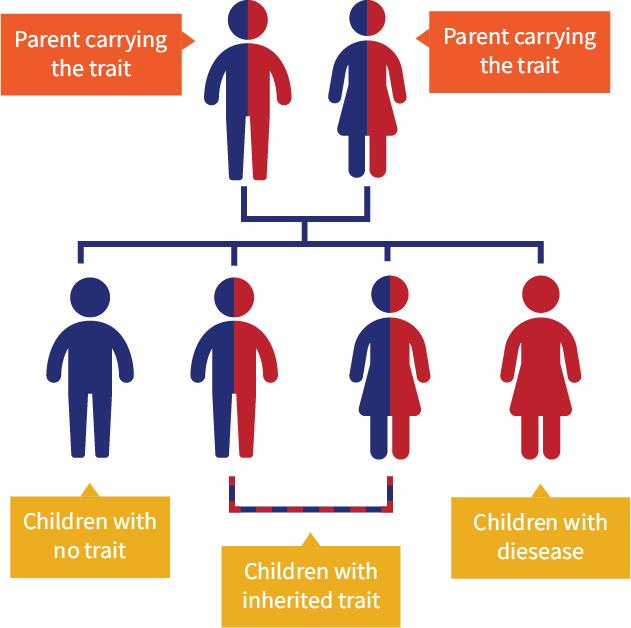
Figure 2. Graphic depicting the genetic path of sickle cell. If each parent possesses the sickle cell trait then roughly there is a 25% chance their child will inherit no copies and not possess the sickle cell trait, a 50% chance that the child will inherit one copy have the sickle cell trait and a 25% chance that their child will inherit both copies and possess sickle cell disease.
SCD is most common among people in Africa and of African descent, but it also can affect Hispanic, Middle Eastern, and Indian people. Roughly 100,000 people in the United States and 14,000 people in the United Kingdom have sickle Cell Disease, with people of African Descent accounting for 90% of cases
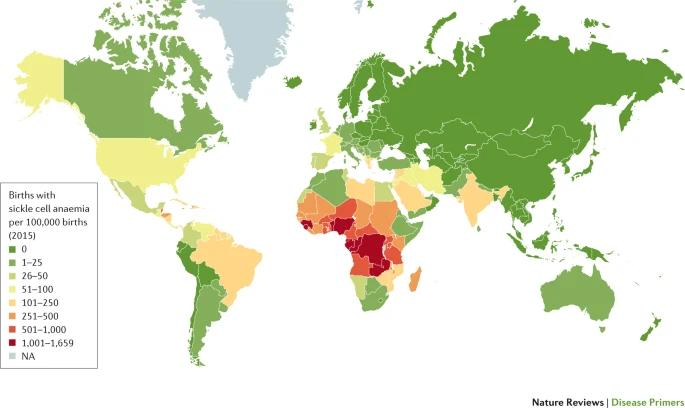
Figure 3. Graphic depicting the number of births with sickle cell anemia for every 100,000 on a world map. Most births with sickle cell anemia are concentrated in West and Central Africa.
Sickle Cell Anemia
Sickle cell anemia, the most common form of SCD, is an autosomal (non-sex cell) recessive blood disorder caused by a point mutation on the hemoglobin beta gene (HBB) on chromosome 11. Hemoglobin is a protein that is the most essential component of red blood cells; it allows oxygen to be delivered to the necessary tissues and organs in the body. A single change in the nucleotide base (adenine, thymine, cytosine, guanine) on the hemoglobin coding gene HBB causes the production of structurally abnormal hemoglobin called HbS, which clusters together misshaping red blood cells into a sickle shape. These disfigured red blood cells struggle to flow through small blood vessels, and a buildup can cause long episodes of severe acute pain called vaso-occlusive crises and eventually damage organs until failure.
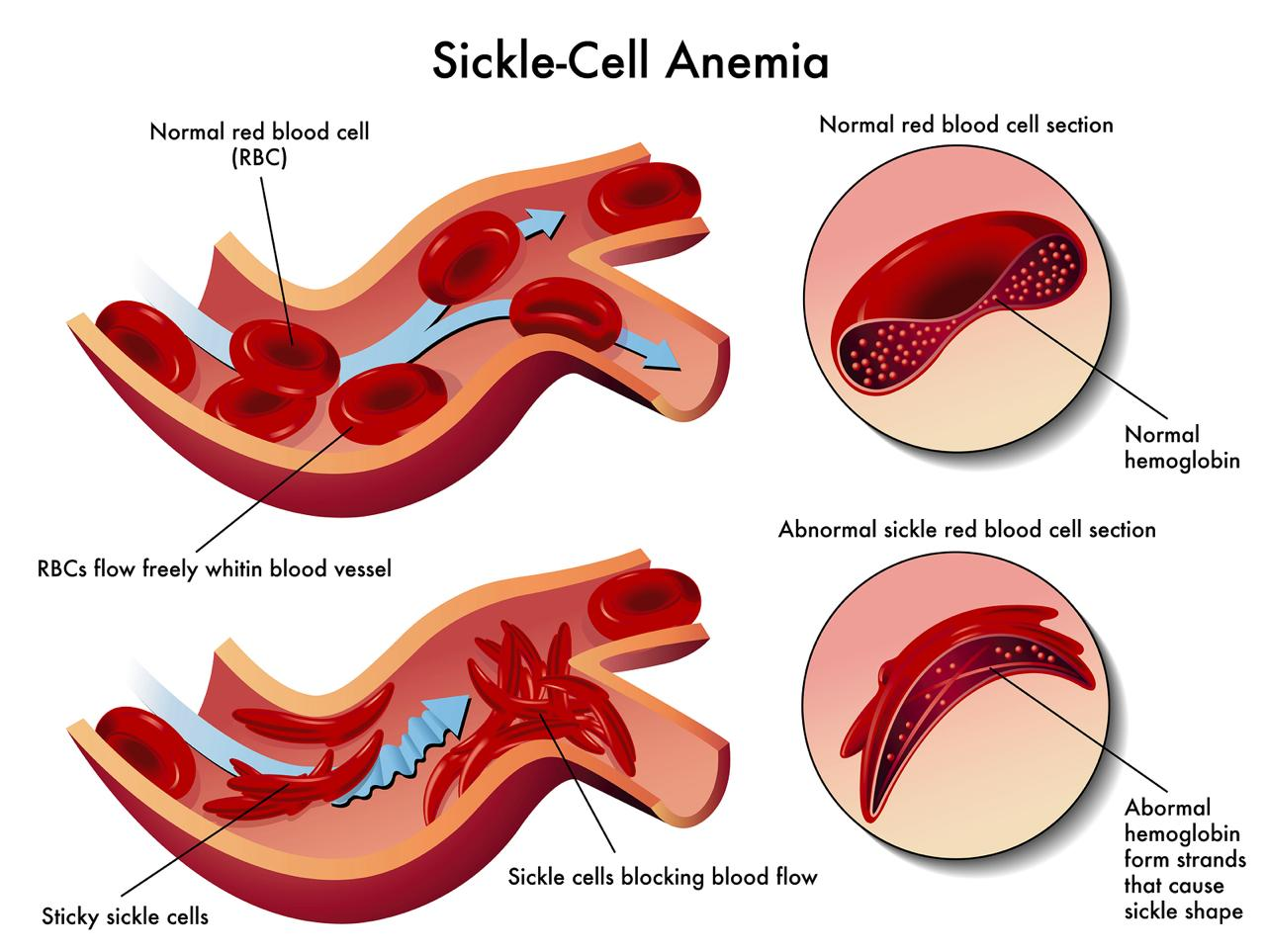
Figure 4. Graphic showing the structure and flow of regular blood cells and sickle cells. Regular blood cells flow freely and efficiently in blood vessels, while sickle cells stick to each other and clog blood vessels.
Bone Marrow Transplantation
Before the recent advent of novel gene therapies, the most effective treatment and known “cure” for SCD was Bone Marrow Transplantation also known as stem cell transplantation. The process replaces faulty red blood cells with stem cells that create healthy red blood cells called hematopoietic stem cells. Although, getting a bone marrow transplant is a several-week arduous journey. First, a donor must be found usually between a sibling or other close relative; however, only 30% of patients are able to find a match within their family and the remaining 70% are left to rely on a national registry for a match with varying percentages of success. After a matched donor has been found, then the patient must go through one to two weeks of chemotherapy to destroy their abnormal red blood cells – with many healthy red blood cells destroyed in the process as well. After the chemo, the donor’s stem cells will be harvested and injected into the patient’s body in a process similar to a blood transfusion. Once the procedure is complete, the replaced bone marrow stem cells should start creating new healthy red blood cells within a month. It will take 6 months or up to a year before the immune system and all the cells of the patient return back to normal healthy levels. In 10% of bone marrow transplant cases, the body rejects the newly implanted stem cells in a disease called graft-versus-host disease and it results in organ damage and often death.
Casgevy
Vertex Pharmaceuticals and CRISPR Therapeutics aimed to change this single and difficult procedure by developing Casgevy: a treatment that functions with CRISPR, a Nobel-prize-winning gene editing technology that allows DNA, the fundamental building block of life, to be edited. To fully understand how Casgevy works, some biological processes must be detailed.
CRISPR stands for Clustered Regularly Interspaced Short Palindromic Repeats and was discovered as a vital component of a bacteria’s immune system in its defense against its endless war with viruses. The CRISPR Cas9 (CRISPR-associated protein) complex acts as “molecular scalpels” directed by a guide RNA that allow for a double-strand cut to be made in DNA to allow for edits, insertions, and deletions.
Casgevy uses CRISPR Cas9 to treat sickle cell by genetically editing DNA to produce fetal hemoglobin and make less adult hemoglobin. As described earlier, hemoglobin is a crucial component of a red blood cell's ability to uptake oxygen, and oxygen affinity varies for fetal hemoglobin (HbF) and adult hemoglobin (HbA). HbF is composed of two alpha and gamma subunits, while HbA is composed of two alpha and beta subunits. Gamma and beta subunits are almost synonymous in structure, except for the fact that the gamma subunit contains a different, neutral, and nonpolar amino acid at position 136, which allows for a conformational change that gives the protein enhanced physiological capabilities in oxygen delivery. Due to its heightened oxygen affinity and delivery over HbA, HbF plays a primary role in fetal development by trapping and transporting oxygen from the mother’s blood to the fetal circulatory system. Briefly after a baby is born, the production of HbF is halted by the BCL11A gene on chromosome two, and by 6 months HbF fully disappears from the blood. For people who have SCD, symptoms start to show when the body starts to produce less HbF and HbA starts to take on a sickle shape.
Casgevy Treatment Process
Casgevy treats SCD by using the CRISPR Cas9 complex to edit the BCL11A gene to suppress its activity so that the body can produce HbF again. The presence of HbF and its high oxygen affinity helps prevent red blood cells from forming a sickle cell shape. The process of receiving Casgevy is fairly simple.
- A patient receives a special medicine called mobilization medicine, which moves blood stem cells from the bone marrow into the bloodstream.
- Then blood is collected from the patient and stem cells are separated out in a process called apheresis. This can take 1 to 2 weeks.
- Then the collected stem cells are taken to a site for precise genetic edits using CRISPR Cas9 on the BCL11A gene.
- These edited stem cells are then returned to the body with a one-time intravenous infusion following the administration of chemotherapy to create space for the new cells.
The recovery process after the transfusion can take four to six weeks. Over time, the edited blood stem cells in the body travel into the bone marrow and start transforming into HbF.
Unlike bone marrow transplantation, this treatment does not need a donor since the patient’s own cells are edited and inserted back into the bloodstream. The treatment also has extremely high efficacy as 93.5% of people in the clinical study did not have severe pain crises for at least a year after treatment, and 100% of people evaluated were not hospitalized for severe pain crises in the same time frame. Casgevy side effects include prolonged low levels of platelets, nausea, and muscle and bone pain. People who have gone through treatment are not allowed to donate blood, organs, tissues, or cells for the rest of their lives, and fertility may be lost.

Figure 5. Graphic depicting the full process of the Casgevy gene-editing treatment. The patient’s stem cells are extracted, edited, and returned back into the blood to create healthy red blood cells Lyfgenia
Developed by Bluebird Bio in Somerville Massachusetts, Lyfgenia is a sickle cell treatment that attacks the disease on the genetic level similar to Casgevy; although, Lyfgenia instead uses a gene-addition technique than a gene-editing one. The process of collecting and filtering blood stem cells from the patient remains the same for both treatments, but the difference lies in the fact that Lyfgenia uses a viral vector to deliver functional copies of the HBB gene in the stem cells before they are returned into the body. Viral vectors are viruses that have been modified and designed to deliver specific genes; Lyfgenia uses a lentivirus, known for causing HIV, as a vector. The HIV-causing aspects of the virus are removed and the efficient genetic delivery mechanisms that the virus possesses are leveraged for directly adding the functional HBB gene to the genome. The treatment is a one-time gene therapy without the need for a donor and boasts high efficacy, with 88% of 32 patients having no pain crises from 6 to 18 months after treatment. Some drawbacks include long platelet recovery time, increased risks of blood cancer, and false positives on HIV tests due to the use of a lentiviral vector. People who have gone through treatment are also not allowed to donate blood, organs, tissues, or cells for the rest of their lives.
Public Health (Accessibility, cost)
As detailed above, Casgevy and Lyfgeny are truly revolutionary therapies that are a great first step in eliminating sickle Cell disease with not too many risks or downsides, except for one small monetary problem: each gene therapy costs $2.2 million and $3.1 million per patient respectively. Due to the intensive procedures of chemotherapy, blood transfusions, blood filtering, gene-edits, and gene additions that each treatment requires, the costs are required to be exorbitant. While the costs are high for these treatments, it must be noted that the price of these one-time treatments may be comparable to or even less than the costs of the ongoing lifelong medical care that sickle cell anemia patients often go through to keep their symptoms manageable. Still, private health plans and programs such as Medicaid, which covers roughly 50% to 60% of sickle cell patients in the US, are struggling with how they can cover the costs of these treatments for the thousands of people that they cover. However, many measures are being taken to ensure that these pricey treatments can be covered for the benefit of patients.
The Centers of Medicare and Medicaid Services, the overseer of Medicaid, proposed a new model that will involve the negotiation of an outcomes-based agreement that will connect the payment of the drug to the specific outcomes. Payment will be linked to the health benefit from the treatment, and if that health benefit isn’t met then drugmakers are forced to reimburse the insurer. Large steps are also being taken in Florida, which has the highest amount of people living with sickle cell disease according to the American Society of Hematology. On June 14th, the Florida Agency for Healthcare Administration voted to cover both gene therapies, which will bring relief to an estimated 7,000 low-income people with subsidized health insurance in Florida. Florida Governor Ron DeSantis also approved a $10 million grant program that will increase access to specialized sickle cell treatments in the coming year. With the novelty and breakthrough potential that the gene therapies Casgevy and Lygenia bring, cooperation between biotechnology companies, insurance agencies, government officials, and physicians is imperative, so that the suffering of thousands across the country can be reduced and potentially make sickle cell a disease of the past.
Ethical Implications and Implications for Other Diseases
Gene-editing with CRISPR has ushered many significant breakthroughs in curing sickle cell. This technology holds the promise for for curing other genetic diseases caused by single mutations such as Hutington’s Disease. Huntington’s disease, similar to sickle cell, is caused by a point mutation in the HTT gene– which helps neurons carry out their normal function. A variation of this gene leads to the adult onset of brutal, cruel, and painful symptoms from twitching uncontrollably to gradually losing the ability to walk, talk, and swallow to an eventual slow and agonizing death. Huntington’s Disease is a rare, but genetically dominant disease that often passes on to children with symptoms of the disease not manifesting until after childbearing years. With these abilities, it is nearly impossible for the disease to die out in the natural evolutionary process.
Fixing Huntington’s disease would be a fairly straightforward genetic edit, and many treatments similar to Casgevy and Lyfgenia could be approved for treating the disease. However, it can be argued that the treatment could be taken even a step further. The current gene therapy treatments for sickle cell work on somatic cells, which are non-reproductive body cells, which means the curative effect of treatments isn’t passed onto children. Offspring with sickle cell disease would have to go through the same treatment (and heavy costs) again. This is distinct from germline editing. The germline refers to the reproductive cells (egg and sperm) and embryonic cells that contain DNA that are passed onto offspring. With genetic edits on the germline, diseases don't just have the potential to be cured from a person, but wholly removed from the human race. Huntington’s Disease is a sneaky disease that serves no purpose or benefit (as opposed to the way the sickle cell mutation provides immunity against malaria), so shouldn’t it be edited in the germline of those afflicted? The same could be said about sickle cell now that there are many measures against malaria today. In a perfect world, the answer would easily be yes since the eradication of these diseases would reduce great suffering in the world, but it is important to realize that we live in an imperfect world, and germline editing can send humanity down a very slippery slope.
As we enter the complex conversation of germline editing, there are a lot of things to consider. Many individuals may reject a “cure” and consider their disease or disability as an integral part of who they are or who they have become. Franklin Delano Roosevelt was diagnosed with infantile paralysis, also known as polio, when he was 39, and despite this, he went on to become the 32nd president of the United States a little over a decade later. His resilience amid adversity made him become a symbol of hope and perseverance for the American people, with his wife Eleanor referring to his disability as a “blessing in disguise.”
Similarly, Miles Davis, one of the most influential jazz musicians in history, can attribute his struggle with sickle cell to giving him resilience and musical inspiration. Renowned painter Vincent Van Gogh and contemporary music artist Kanye West, both of whom have dealt with Bipolar disorder, can also attribute the disorder to giving them the creativity and vibrant mind to produce art and music that has significantly influenced the world. This begs the question: would the eradication of these “afflictions” completely erase extraordinary creativity, resilience, and genius from humans?
Conversely, many would still push for the elimination of these diseases regardless of some of the added benefits, especially when you consider that these diseases led to substance abuse, death, and heavy controversy from the aforementioned people. However, the full elimination of these diseases leads to an even bigger ethical conversation: should we take another step in our genetic editing capabilities to not just treat, but rather enhance human traits such as intelligence, height, and strength, or even introduce special blood cells that can triple oxygen flow efficiency within the body? These genetic modifications do not necessarily address a problem or disease, but if they contribute to human betterment, are they morally justifiable? Who would control who gets enhancements, the government or the people? Would this increase inequality and even encode it in our genome? Is it better to leave random genetic mutations to nature’s lottery or put it in our hands? Would be “playing God” or just using the tools God bestowed on us?
As we approach the era of “designer babies” there are many questions to consider, but it is crucial to proceed with caution and ethical consideration. The potential for profound advancements exists, but it is imperative that we navigate this new frontier with the utmost care and responsibility. The genie has already escaped the bottle when in November 2018 a Chinese biophysicist decided to create the first “designer babies” by genetically editing twins when they were embryos to become resistant to the HIV virus. Although he was met with much horror, shock, and a prison sentence, it is only a matter of time before acceptance sets in as did with the biological feats of cloning and in-vitro fertilization in the late 20th century. More detailed research, worldwide symposiums, and a deliberation of what values we as humans hold dear must happen before we conduct germline editing again regardless of the aspect or purpose.
Conclusion
FDA-approved gene therapies Casgevy and Lyfgenia represent a pioneering step forward in the treatment of widespread genetic diseases. These therapies have already brought significant relief to many patients facing sickle cell disease, and potential still exists for treating conditions such as Alzheimer’s, blindness, and even cancer. As researchers and biotechnology companies continue to explore the capabilities of groundbreaking gene-editing tools, it's essential to apply prudence and sound judgment. These powerful and promising technologies have the power to fundamentally alter our species in a positive or negative way, so careful and ethical application is crucial going forward.
References:
Bauer, D. E., & Orkin, S. H. (2015). Hemoglobin switching’s surprise: the versatile transcription factor BCL11A is a master repressor of fetal hemoglobin. Current Opinion in Genetics & Development, 33, 62–70. https://doi.org/10.1016/j.gde.2015.08.001
Data and statistics on sickle cell disease. (2024, May 15). Sickle Cell Disease (SCD). https://www.cdc.gov/sickle-cell/data/index.html
Eggert, B. N. (2018, March 13). Tracing sickle cell back to one child, 7,300 years ago. https://www.bbc.com/news/world-africa-43373247
FDR and Polio - FDR Presidential Library & Museum. (n.d.). https://www.fdrlibrary.org/polio
Fu, Y., Andemariam, B., & Herman, C. (2023). Estimating sickle cell disease prevalence by state: A model using US-Born and Foreign-Born State-Specific population data. Blood, 142(Supplement 1), 3900. https://doi.org/10.1182/blood-2023-189287
History & timeline of SCD — Sickle Cell Foundation of Minnesota. (n.d.). Sickle Cell Foundation of Minnesota. https://www.sicklecellmn.org/scdhistory
How CASGEVY works. (n.d.). https://www.casgevy.com/sickle-cell-disease/how-casgevy-works
Kaufman, D. P., Khattar, J., & Lappin, S. L. (2023, March 20). Physiology, fetal hemoglobin. StatPearls - NCBI Bookshelf. https://www.ncbi.nlm.nih.gov/books/NBK500011/
Kurzgesagt – In a Nutshell. (2016, August 10). Genetic engineering will change everything forever – CRISPR [Video]. YouTube. https://www.youtube.com/watch?v=jAhjPd4uNFY
MacMillan, C. (2023, December 19). Casgevy and Lyfgenia: two gene therapies approved for sickle cell disease. Yale Medicine. https://www.yalemedicine.org/news/gene-therapies-sickle-cell-disease
Pagliarulo, N. (2024, January 31). New CMS pilot to test payment scheme for pricey sickle cell gene therapies. BioPharma Dive. https://www.biopharmadive.com/news/medicaid-sickle-cell-gene-therapy-pilot-outcomes-payment/706079/
Pulrang, A. (2024, February 27). What do disabled people mean when we say we don’t want a cure? Forbes. https://www.forbes.com/sites/andrewpulrang/2024/02/26/what-do-disabled-people-mean-when-we-say-we-dont-want-a-cure/
Smith, M. (2024, March 17). Bone marrow transplants for sickle cell disease. WebMD. https://www.webmd.com/a-to-z-guides/bone-marrow-transplant-sickle-cell
Sickle cell disease. (n.d.). Hematology.org. https://www.hematology.org/education/patients/anemia/sickle-cell-disease
The need for more marrow donors | Blood stem cell. (2024, March 1). Blood Stem Cell. https://bloodstemcell.hrsa.gov/donor-information/donate-bone-marrow/need-more-marrow-donors
Thein, S. (2011). Abnormalities of the structure and synthesis of hemoglobin. In Elsevier eBooks (pp. 131–155). https://doi.org/10.1016/b978-0-7020-3147-2.00009-2
Image References
Figure 1:
Zamecnik, A., & Zamecnik, A. (2023, October 26). How gene therapies can transform sickle cell disease treatment. Pharmaceutical Technology. https://www.pharmaceutical-technology.com/features/how-gene-therapies-can-transform-sickle-cell-disease-treatment/
Figure 2:
What is sickle cell trait? — Massachusetts Sickle Cell Association. (n.d.). Massachusetts Sickle Cell Association. https://sicklecellhope.org/what-is-sickle-cell-trait
Figure 3:
Kato, G. J., Piel, F. B., Reid, C. D., Gaston, M. H., Ohene-Frempong, K., Krishnamurti, L., Smith, W. R., Panepinto, J. A., Weatherall, D. J., Costa, F. F., & Vichinsky, E. P. (2018). Sickle cell disease. Nature Reviews. Disease Primers, 4(1). https://doi.org/10.1038/nrdp.2018.10
Figure 4:
MEd, J. H. M. (2018, January 1). Laboratory evaluation of sickle cell disease in the ED — taming the SRU. Taming the SRU. https://www.tamingthesru.com/blog/intern-diagnostics/laboratory-evaluation-of-sickle-cell-disease-in-the-ed
Figure 5:
Walsh, B. F. (2023, November 16). Casgevy: UK approves gene-editing drug for sickle cell. https://www.bbc.com/news/health-67435266
See More Posts
Copyright © 2021 Govest, Inc. All rights reserved.


